
PASQAL has successfully implemented for the first time an algorithm on a commercial neutral atoms quantum device to tackle a critical molecular biology problem in drug discovery. This project has been carried out in collaboration with Qubit Pharmaceuticals.
Proteins are large molecules that play critical roles in our bodies, such as helping build and repair tissues, drive metabolic reactions, and maintain fluid balance. However, because proteins are responsible for such essential processes, they can also be harmful. If something goes wrong while a protein is being built in a cell, the result could be a toxic protein. For example, cancers are related to modifications occurring at the DNA level, producing toxic proteins and modifying the capacity of cells to operate normally. In fact, faulty proteins account for most of the world’s sickness.
Drugs that may help fight diseases are often simpler, smaller molecules, called ligands, designed to get attached to a strategic place in the toxic protein to inhibit its harmful action.
To design a medicine, researchers need to investigate the binding process between the drug and the targeted harmful protein. Because proteins live and function inside cells, the drug-protein binding process must be understood in an environment filled with the most common substance in the cell: water. In fact, water can impact dramatically the way a targeted protein interacts with a drug.
Hunting for water molecules inside proteins
Proteins are long chains of molecules called amino acids that interact with each other, causing the chain to fold — like a ribbon — creating complex structures that may have pockets. The water in the cell penetrates the inner part of the protein, filling its pockets, affecting the size and scaffold of the protein, and mediating the interaction between a protein and a ligand. Investigating where and how many water molecules a protein may have in its pockets is crucial for designing medicines able to inhibit the toxic behavior of the targeted protein.
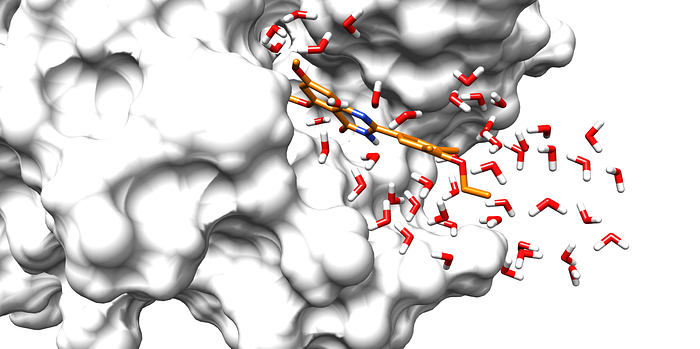
Computational methods for understanding protein hydration have significantly advanced alongside experimental approaches, saving time and reducing costs, facilitating drug discovery processes. Moreover, in most cases, computational techniques can provide a better view of the structure and dynamics of the system than experimental setups. The reason is that crowded areas and inner cavities in a protein require very high resolution, while X-ray crystallographic experiments can only provide limited information due to poor resolution.
Numerical computational techniques can be very helpful. They can be used to simulate the explicit presence and interaction of water molecules in a protein. However, these simulations are usually costly, and the time required to provide accurate predictions can be extremely long, mainly if the cavity under investigation is occluded enough.
An alternative approach is first finding the water density in the protein pockets and then extracting the water molecules’ position from the density.
However, the number of configurations — the different ways water molecules can be placed in a pocket — corresponding to a given density remains potentially extremely large for classical methods.
A hybrid quantum/classical algorithm to locate water molecules inside protein pockets
Quantum computing is nowadays a great source of hope to deal with problems that are too complex for classical computers. By leveraging the principles of quantum physics, quantum processing units can store loads of information simultaneously and perform exceptionally well in tackling problems with a large number of combinations. See examples in previous blogs here and here.
PASQAL, in collaboration with Qubit Pharmaceuticals, is developing a hybrid quantum/classical approach that uses a classical algorithm to find the water density information in the protein and then a quantum algorithm to locate the water molecules inside any pocket, even in the buried ones. The collaboration will implement this quantum/classical water placement algorithm in a PASQAL quantum device.
Experimental test on Fresnel, a PASQAL commercial quantum processing unit
Scientists at PASQAL successfully tested a preliminary version of the novel quantum water placement algorithm on Fresnel 1, the first PASQAL industrial neutral atoms quantum computer. For this proof of concept, the scientists used the Major UrinaryProtein-1 (MUP-1), a protein produced predominantly from the liver. Evidence shows that this protein plays a major role in communication through pheromones, and in regulating energy expenditure and metabolism.
The following figure shows that the experimental results (on the right) are in agreement with the theoretical predictions emulations (on the left). Each figure represents a 2D slice in the protein pocket where the yellow shade represents the water placement.
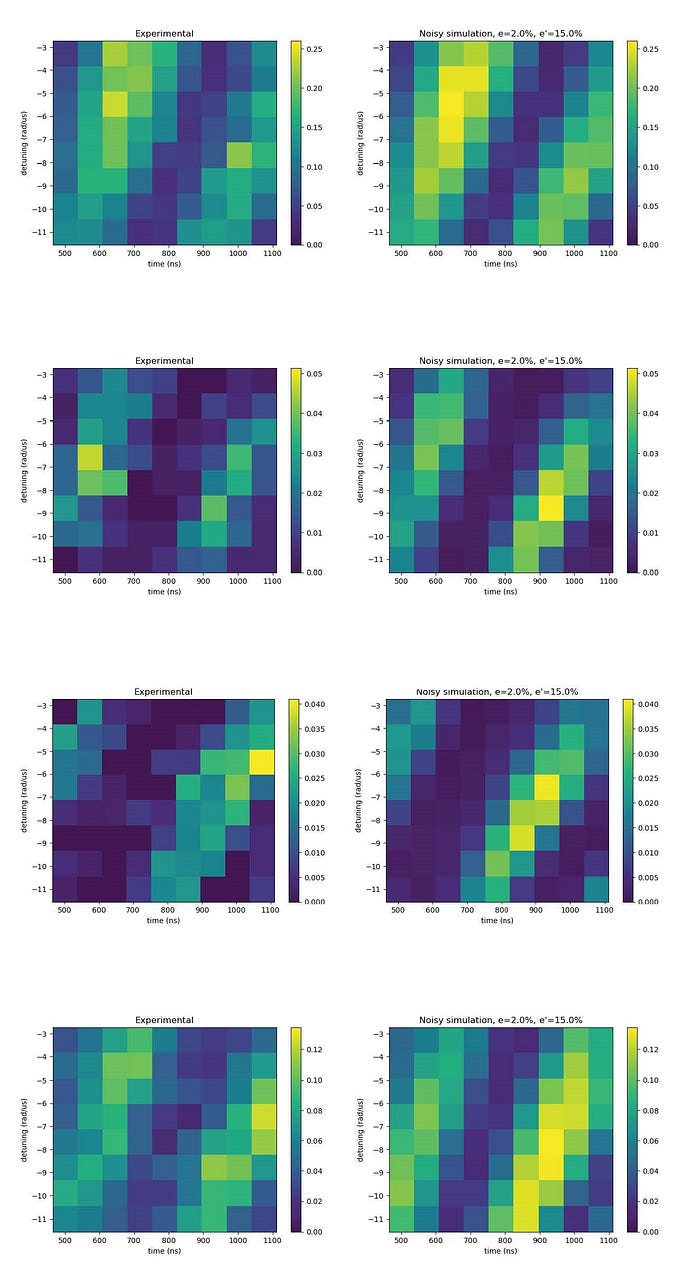
This successful result indicates that meaningful solutions to the water placement problem can be obtained using neutral atom devices, such as PASQAL’s. A preprint with these results has been recently published in the arxiv. This is the first time such an experiment has been conducted with the use of a real quantum computer, showing the capacity of quantum technologies to contribute to advancing healthcare problems. The full version of the PASQAL water placement algorithm will be implemented on the next-generation neutral atom machine that will be operating with 1000 qubits.
A quantum algorithm to accelerate drug discovery explained
PASQAL quantum algorithm uses the density of water inside the protein pockets, which is calculated with a classical computer using a method called 3D Reference Interaction Site Model (3D-RISM). The3D-RISM algorithm produces a probability distribution of densities, which is sort of a continuous and structureless pattern, like blur stains on a piece of paper. The question is, how do we extract the position of each water molecule from that messy output?
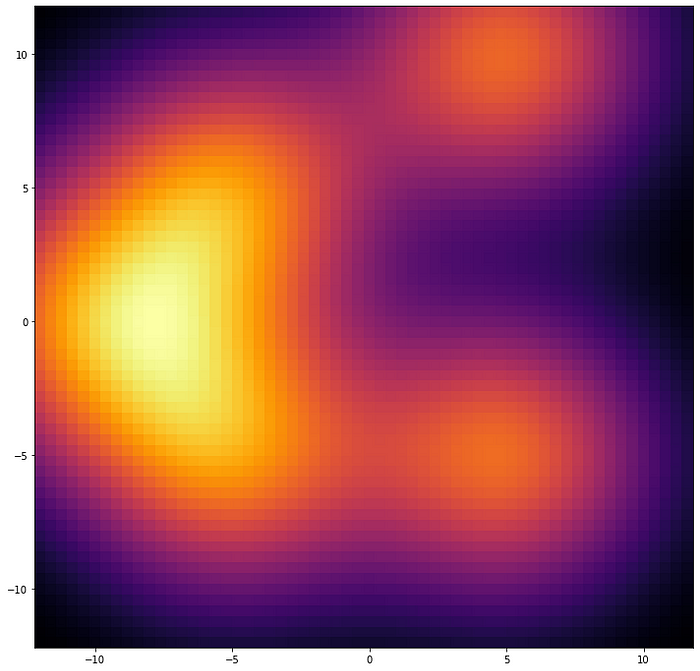
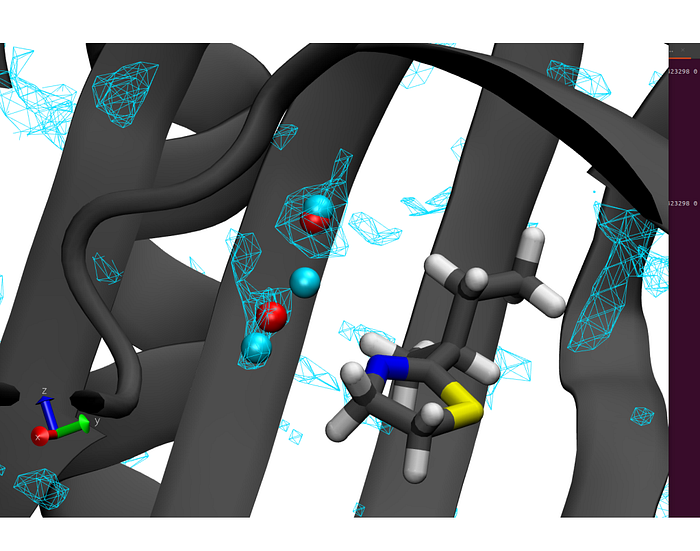
For this task, PASQAL and Qubit Pharmaceuticals have created a quantum algorithm that will start with the opposite question: if we propose a distribution of waters, does it reproduce the density map given by the 3D Reference Interaction Site Model?
The first step is to guess the positions of the water molecules assuming high water density places, each position with a probability distribution. In other words, we “smear” them so that they look like a blurry, spread-out continuous distribution. Then compare these blurry configurations with the output provided by the 3D Reference Interaction Site Model using a quantity representing a “distance.” The idea is to produce as many combinations as possible to find the minimal “distance” between these two density patterns.
To minimize this distance, we use the fact that, in real life, the water molecules are interacting, creating networks through hydrogen bonds, and that there is a minimal physical distance between each other.
PASQAL quantum method is inspired by the best classical software created for this job called Gasol. However, Gasol assigns a penalty included artificially to configurations where two water molecules would be placed too close to each other. This constraint is naturally implemented with neutral atoms architectures, let’s see how.
Neutral atoms quantum devices use highly focused lasers, so-called optical tweezers, to trap and manipulate neutral atoms individually to create 1D, 2D, and 3D arrays in arbitrary configurations. In these architectures, each qubit is represented by a two-level atomic energy state, usually a ground state and a Rydberg state, a very high energy state. In the Rydberg state, atoms are polarized, inducing van der Waals’ interactions between them.
This Van der Waals-type interaction between the atoms forbids two atoms to be in the Rydberg state simultaneously, if they are too close to each other, a phenomenon called Rydberg blockade.
In the experiment conducted on Fresnel 1, we used 2D arrays of rubidium atoms in the Rydberg state to represent the position and probability distribution of water molecules inside a protein pocket, using the Rydberg blockade as a natural way to keep waters being too close to each other during the simulation.
The future of quantum computer-aided drug design
Much of a drug discovery project’s efforts are put in identifying and studying the pocket to find and optimize a drug with high affinity to the protein target.
Despite several successes, it is still extraordinarily difficult to predict binding affinity between a protein and a ligand. The ability to design novel ligands at will that inhibit the action of harmful biomolecules remains one of the major challenges in contemporary health sciences. The successful implementation of a quantum algorithm that helps tackle protein ligand affinity would represent a tremendous step further toward improving our quality of life.
References
D’Arcangelo,M., Loco, D., Fresnel team, et al. (2023). Leveraging Analog Quantum Computing with Neutral Atoms for Solvent Configuration Prediction in Drug Discovery. Preprint available here: https://arxiv.org/abs/2309.12129.
Henriet, L. et al. (2020). Quantum computing with neutral atoms. Quantum, 4, 327.
Samways, M. L. et al. (2021).Water molecules at protein–drug interfaces: computational prediction and analysis methods. Chem. Soc. Rev., 50(16), 9104–9120.
Bucher, D. et al.(2018). Shedding Light on Important Waters for Drug Design: Simulations versus Grid-Based Methods. J. Chem. Inf. Model., 58(3), 692–699.
Wlodawer, A. et al. (2008). Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. FEBS J., 275(1), 1–21.
Michel, J. et al.(2009). Prediction of the Water Content in Protein Binding Sites. J. Phys. Chem. B,113(40), 13337–13346.
Baron, R. et al.(2010). Water in Cavity−Ligand Recognition. J. Am. Chem. Soc.,132(34), 12091–12097.
Fusani, L. et al.(2018). Optimal water networks in protein cavities with GAsol and 3D-RISM. Bioinformatics,34(11), 1947–1948.
The full water placement inside proteins project will be funded by the Quantum for Bio program. Launched in spring 2023 by Wellcome Leap, an offshoot of the Wellcome Trust, the Quantum for Bio program aims to accelerate the use of quantum computing in healthcare by developing applications that will benefit from the arrival of quantum computers within 3–5years.
Would you like to learn more about these techniques on a neutral atom quantum computer? Get familiar with quantum computing, our platform, and algorithms with Quantum Discovery.